
Hubungan Antara Delesi 22q11.2 dengan Tetralogy of Fallot

Penulis: dr. Gema Citra Dwijayanti
Tetralogy of Fallot (TOF) merupakan penyakit jantung kongenital (sianotik) terbanyak dari seluruh penyakit jantung bawaan sianotik dan mencakup 7% dari seluruh kelainan jantung yang ada.(Mercer-Rosa et al., 2020) 22q11.2 deletion syndrome (Di George syndrome) merupakan sindrom genetika yang disebabkan delesi dari segmen kecil pada kromosom 22 yang memiliki gambaran klinis bervariasi seperti adanya kelainan jantung bawaan, wajah dismorfik, kejadian infeksi berulang, developmental delay, gangguan perkembangan dan cleft palate, Di George Syndrome juga merupakan kelainan kromosom yang paling sering ditemukan pada pasien dengan TOF atau PA (Pulmonary atresia) / VSD (Ventricle Septal Defect) dan berhubungan dengan peningkatan angka morbiditas dan mortalitas yang tinggi dibandingkan dengan pasien tanpa delesi 22q11.2. (Kauw et al., 2020).
Sekitar 15% pasien dengan Tetralogy of Fallot (TOF) memiliki delesi pada 22q11.2. (Momma et al., 2001) Delesi 22q11.2 pada pasien Tetralogy of Fallot berhubungan dengan kejadian anomali pada arkus aorta dan percabangannya, a. pulmonalis dan ductus arteriosus. Pada penelitian yang dilakukan baru-baru ini, delesi 22q11.2 berhubungan dengan prognosis yang buruk paska dilakukan operasi koreksi TOF, termasuk pemanjangan waktu lama rawat di RS dan ICU, peningkatan lama penggunakan ventilator dan peningkatan jenis obat-obatan yang digunakan dalam terapi.(Kauw et al., 2020). Terdapat kelainan ekstra kardiak yang disebabkan oleh 22q11.2 deletion syndrome, antara lain wajah dismorfik, velopharyngeal insufficiency, hypocalcaemia, renal anomalies, immune deficiencies, dan mental retardation. Pada pasien TOF atau PA/VSD yang memiliki delesi pada 22q11.2 perlu dilakukan stratifikasi risiko dan konseling genetika. (Mercer-Rosa et al., 2015)
Mekanisme 22q11.2 deletion syndrome-TOF
Kelainan pada jantung muncul pada 80% neonatus yang memiliki 22q11.2 deletion syndrome. Terdapat 3 gen (TBX1, CRKL, dan ERK2) pada Chromosome 22q11.2 yang telah diidentifikasi merupakan penyebab terjadinya disfungsi pada sel-sel neural crest dan bagian anterior jantung serta anomali dari 22q11.2 deletion syndrome. Kelainan tersering adalah anomali conotruncal, dimana gangguan pada conotruncal dapat menyebabkan Tetralogy of Fallot (TF), TF dengan pulmonary atresia, truncus arteriosus, serta interupsi arkus aorta. (Momma et al., 2010)
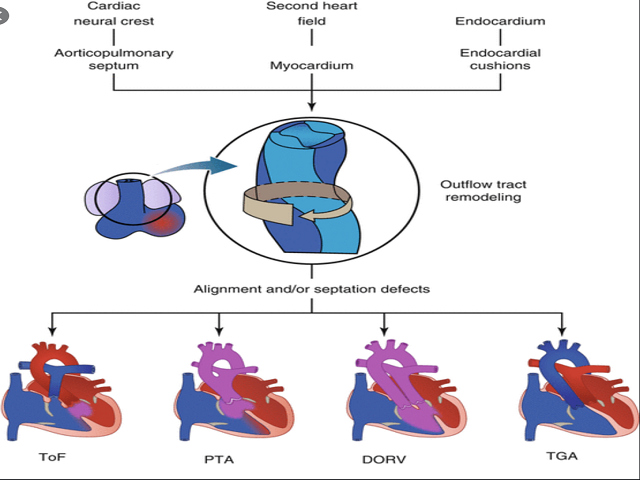
Gambar 1. Anomali Conotruncal (Laforest B., Zaffran S, 2016)
Anomali Conotruncal merupakan malformasi pada infundibulum (conus arteriosus) dan aorta (truncus arteriosus) yang memiliki kelainan ventriculo-arterial alignments dan koneksi: tetralogy of Fallot, truncus arteriosus, interrupted aortic arch type B, transposition of the great arteries, double-outlet right ventricle, double-outlet left ventricle, anatomically corrected malposition of the great arteries. (Van Praagh R., 2016)
Referensi :
Ben-Shachar, S., Ou, Z., Shaw, C., Belmont, J., Patel, M., Hummel, M., Amato, S., Tartaglia, N., Berg, J., Sutton, V., Lalani, S., Chinault, A., Cheung, S., Lupski, J. and Patel, A., 2008. 22q11.2 Distal Deletion: A Recurrent Genomic Disorder Distinct from DiGeorge Syndrome and Velocardiofacial Syndrome. The American Journal of Human Genetics, 82(1), pp.214-221.
Kauw, D., Woudstra, O., van Engelen, K., Meijboom, F., Mulder, B., Schuuring, M. and Bouma, B., 2020. 22q11.2 deletion syndrome is associated with increased mortality in adults with tetralogy of Fallot and pulmonary atresia with ventricular septal defect. International Journal of Cardiology, 306, pp.56-60.
Laforest B., Zaffran S. (2016) Genetics of Conotruncal Anomalies. In: Lacour-Gayet F., Bove E., Hraška V., Morell V., Spray T. (eds) Surgery of Conotruncal Anomalies. Springer, Cham. https://doi.org/10.1007/978-3-319-23057-3_36
Mercer-Rosa, L., Paridon, S., Fogel, M., Rychik, J., Tanel, R., Zhao, H., Zhang, X., Yang, W., Shults, J. and Goldmuntz, E., 2015. 22q11.2 Deletion Status and Disease Burden in Children and Adolescents With Tetralogy of Fallot. Circulation: Cardiovascular Genetics, 8(1), pp.74-81.
